Mechanically Generated Friction Fits in Power Transmission Couplings
Mechanically Generated Friction Fits in Power Transmission Couplings

In the world of electric motors and power transmission coupling products, torque is increasingly being transferred between directly coupled shafts by means of purely mechanically generated friction rather than by positive drive connections like keys and keyways or splines. Mechanical friction fits evolved into modern standardized dimensions during the twentieth century as machine tools and cutting tooling evolved into their present states of precision and accuracy. For many decades power transmission component design lagged behind the machining world when it came to the use of mechanically generated frictional clamping, with a large portion of connecting elements still relying on keyways and interference fits to guarantee transmission.
Interference fits, wherein the diameter of the male component is actually slightly larger than the diameter of the mating female component, do have their merits when it comes to ensuring reliable backlash free transmission. But they can be difficult to assemble, as the shaft must be cooled with a cryogenic such as liquid nitrogen, or the bore needs to be expanded with heat. If either high heat or extreme cold are used, safety is a key concern during the assembly process. Heating a metal component will also often change its mechanical properties such as temper, so cooling the male fitting part is preferred when maintaining the material properties is a concern. Due to the inconveniences associated with interference fits, most newly designed frictional clamping coupling hubs are made using compression by means of screw thread fasteners instead.
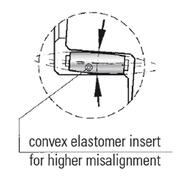
This type of mechanical connection is very  predictable and can be made by applying a set amount of torque to radial or axial screws. For easy assembly the fit between the male and female part typically should not have overlapping diametrical tolerances, but should also not allow for excessive clearance. When the fits can be made by hand, proper tightening of the fasteners is all that is necessary to create a friction fit which will reliably and predictably transmit torque for the life of the machine. A shrink disk style fit on a female member of a coupling will maintain concentricity and will compensate for tolerances on a male shaft while maintaining the ability to transmit torque across a range of diameter dimensions and torque values. This type of conical clamping hub can be manufactured in different styles to promote modularity, ease of assembly and concentricity between shafts, often allowing for easier balancing for high speed applications.
predictable and can be made by applying a set amount of torque to radial or axial screws. For easy assembly the fit between the male and female part typically should not have overlapping diametrical tolerances, but should also not allow for excessive clearance. When the fits can be made by hand, proper tightening of the fasteners is all that is necessary to create a friction fit which will reliably and predictably transmit torque for the life of the machine. A shrink disk style fit on a female member of a coupling will maintain concentricity and will compensate for tolerances on a male shaft while maintaining the ability to transmit torque across a range of diameter dimensions and torque values. This type of conical clamping hub can be manufactured in different styles to promote modularity, ease of assembly and concentricity between shafts, often allowing for easier balancing for high speed applications.
The fastener assisted friction fit has streamlined design of  couplings as it allows for the use of smooth shafts which are not subject the imbalance and complications shaft key or spline tolerances can cause. The next time you are sizing and selecting rotating components, consider going with keyless frictional fits and reap the benefits of modern machine construction.
couplings as it allows for the use of smooth shafts which are not subject the imbalance and complications shaft key or spline tolerances can cause. The next time you are sizing and selecting rotating components, consider going with keyless frictional fits and reap the benefits of modern machine construction.











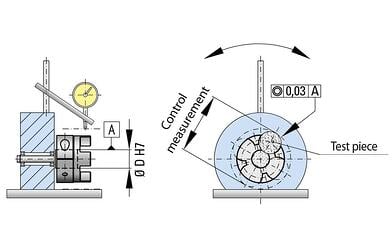
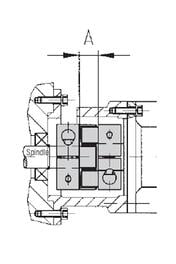
 (self-centering clamping system)
(self-centering clamping system)
















 One of the oldest methods mechanical designers have used to damp vibrations is employing a flywheel. This device is still widely in use, in many parts of industry. Using laws of basic classical mechanics, “a body in motion tends to stay in motion, while a body at rest tends to stay at rest,” designers know that it is difficult to change the rotating velocity of a massive, high inertia flywheel easily. This is why a flywheel is used on the output shaft of many combustion engines, since they tend to continue rotating smoothly rather than jerking severely every time a cylinder fires. A large drive coupling or belt pulley would have the same effect. Piston air compressors often have massive cast iron sheaves driving them for this reason.
One of the oldest methods mechanical designers have used to damp vibrations is employing a flywheel. This device is still widely in use, in many parts of industry. Using laws of basic classical mechanics, “a body in motion tends to stay in motion, while a body at rest tends to stay at rest,” designers know that it is difficult to change the rotating velocity of a massive, high inertia flywheel easily. This is why a flywheel is used on the output shaft of many combustion engines, since they tend to continue rotating smoothly rather than jerking severely every time a cylinder fires. A large drive coupling or belt pulley would have the same effect. Piston air compressors often have massive cast iron sheaves driving them for this reason.